
Breaking the tape
The top performers among drugs launched in 2020 were each the first of their kind.
By Joshua Slatko • [email protected]
The leaders in pharma’s Class of 2020 were all firsts. Veklury, for COVID, and Tepezza, for thyroid eye disease, were each the first drug of any kind to be approved by FDA for their respective disease targets. And while other treatments for migraine exist, Ubrelvy was the first orally administered calcitonin gene-related peptide (CGRP) receptor antagonist (gepant) for the treatment of migraine attacks once they start. The era of follow-ons in pharma may not entirely be over; but surely the industry’s researchers are still breaking barriers.
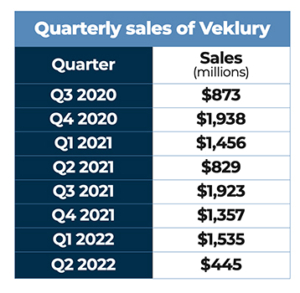
Earning nearly $5.57 billion in sales during the product’s first full calendar year on the market, Veklury was the first drug approved by FDA for the treatment of COVID-19.
Veklury
The very first drug to be approved in the United States for the treatment of COVID-19, Gilead’s Veklury received emergency use authorization from FDA during May 2020, an expanded EUA three months later, and full approval for treating patients with COVID requiring hospitalization during October 2020. Veklury had originally been developed for the treatment of hepatitis C and had been studied in Ebola and Marburg virus, without success.
FDA approval was based on three randomized controlled trials including final results of the National Institute of Allergy and Infectious Diseases’ double blind, placebo-controlled Phase III ACTT-1 trial, which showed that treatment with Veklury resulted in clinically meaningful improvements across multiple outcome assessments compared with placebo in hospitalized patients with COVID-19. Based on the strength of these data, Veklury became a standard of care for the treatment of COVID-19 in hospitalized patients.
In the randomized, double-blind, placebo-controlled ACTT-1 trial, Veklury significantly improved time to recovery as compared to placebo – by five days in the overall study population (10 versus 15 days) and seven days in patients who required oxygen support at baseline (11 versus 18 days). As a secondary endpoint, Veklury also reduced disease progression in patients needing oxygen, resulting in a significantly lower incidence of new mechanical ventilation or ECMO (13 percent versus 23 percent). In the overall patient population, there was a trend toward reduced mortality with Veklury compared with placebo at Day 29.
In June 2021, Gilead announced positive data from three retrospective studies of the real-world treatment of patients hospitalized with COVID-19, adding to the body of mortality and hospital discharge data for patients treated with Veklury. All three of the real-world analyses observed that, in the overall patient populations, patients who received Veklury treatment had significantly lower risk for mortality compared with matched controls. A reduction in mortality was observed across a spectrum of baseline oxygen requirements. The results were consistently observed at different time frames over the course of the pandemic and across geographies. Two of the studies also observed that patients who received Veklury had a significantly increased likelihood of discharge from the hospital by Day 28.
In January 2022, FDA granted expedited approval of a supplemental new drug application for Veklury for the treatment of non-hospitalized adult and adolescent patients who are at high risk of progression to severe COVID-19, including hospitalization or death. The expanded indication allowed for Veklury to be administered in qualified outpatient settings that can administer daily intravenous infusions over three consecutive days. FDA also expanded the pediatric EUA of Veklury to include non-hospitalized pediatric patients younger than 12 years of age who are at high risk of disease progression.
These actions by FDA came amidst a surge in COVID-19 cases and the reduced susceptibility to several anti-SARS-CoV-2 monoclonal antibodies (mAbs) due to the Omicron variant. In contrast, Veklury targets the highly conserved viral RNA polymerase, thereby retaining activity against existing SARS-CoV-2 variants of concern. In vitro laboratory testing has shown that Veklury retains activity against the Omicron variant.
 The FDA sNDA approval, pediatric EUA expansion, and updated National Institutes of Health Treatment Guidelines for COVID-19 that additionally recommend Veklury for treatment in non-hospitalized settings were based on results from the PINETREE Phase III randomized, double-blind, placebo-controlled trial. The study evaluated the efficacy and safety of a three-day course of Veklury for intravenous use for the treatment of COVID-19 in non-hospitalized patients at high risk for disease progression. An analysis of 562 participants randomly assigned in a 1:1 ratio to receive Veklury or placebo, demonstrated that treatment with Veklury resulted in a statistically significant 87 percent reduction in risk for the composite primary endpoint of COVID-19 related hospitalization or all-cause death by Day 28 (0.7 percent, 2/279) compared with placebo (5.3 percent, 15/283). In the study, no deaths were observed in either arm by Day 28.
The FDA sNDA approval, pediatric EUA expansion, and updated National Institutes of Health Treatment Guidelines for COVID-19 that additionally recommend Veklury for treatment in non-hospitalized settings were based on results from the PINETREE Phase III randomized, double-blind, placebo-controlled trial. The study evaluated the efficacy and safety of a three-day course of Veklury for intravenous use for the treatment of COVID-19 in non-hospitalized patients at high risk for disease progression. An analysis of 562 participants randomly assigned in a 1:1 ratio to receive Veklury or placebo, demonstrated that treatment with Veklury resulted in a statistically significant 87 percent reduction in risk for the composite primary endpoint of COVID-19 related hospitalization or all-cause death by Day 28 (0.7 percent, 2/279) compared with placebo (5.3 percent, 15/283). In the study, no deaths were observed in either arm by Day 28.
In February, Gilead released data demonstrating the in vitro activity of Veklury against 10 SARS-CoV-2 variants, including Omicron. Results of Gilead’s studies were consistent with other in vitro studies independently conducted by researchers from institutions in other countries, including Belgium, the Czech Republic, Germany, Poland and the United States, which confirmed Veklury’s antiviral activity against multiple previously identified variants of SARS-CoV-2, including Alpha, Beta, Gamma, Delta and Omicron.
The study analyzed in vitro antiviral activity by two methods to understand the susceptibility of 10 major SARS-CoV-2 variants to Veklury. The study results showed similar activity of Veklury against the variants and an early ancestral A lineage isolate detected in Seattle, Wash. (WA1 strain). Specifically, Delta and Omicron variants both remained fully susceptible to Veklury, and these laboratory results demonstrated that Veklury has remained active against all major variants isolated over the past two years.
In April, FDA approved a supplemental new drug application for Veklury for the treatment of pediatric patients who are older than 28 days, weighing at least 3 kg, and are either hospitalized with COVID-19 or have mild-to-moderate COVID-19 and are considered high risk for progression to severe COVID-19, including hospitalization or death. This approval made Veklury the first and only approved treatment for pediatric COVID patients in the United States. Under the expanded indication, a three-day Veklury treatment regimen is recommended to help prevent hospitalization in non-hospitalized COVID-19 pediatric patients who are at high risk for COVID-19 disease progression. For hospitalized pediatric patients who do not require invasive mechanical ventilation and/or ECMO, a five-day treatment course is recommended. The approval was supported by results from the CARAVAN Phase II/III single arm, open-label study, which demonstrated that Veklury was generally well-tolerated among pediatric patients hospitalized with COVID-19 with a high proportion of participants showing clinical improvement and recovery, as well as data from trials in adults.
Tepezza

Tepezza was the first drug ever approved by FDA for the treatment of thyroid eye disease.
When it earned approval in January 2020, Horizon Therapeutics’ Tepezza became the first and only FDA-approved medicine for thyroid eye disease, a serious, progressive and vision-threatening rare autoimmune disease that is associated with proptosis (eye bulging), diplopia (double vision), blurred vision, pain, inflammation, and facial disfigurement. Tepezza is a fully human monoclonal antibody (mAb) and a targeted inhibitor of the insulin-like growth factor-1 receptor (IGF-1R) that is administered to patients once every three weeks for a total of eight infusions.
The FDA approval of Tepezza was supported by a robust body of clinical evidence, including statistically significant, positive results from the Phase II clinical study, as well as the Phase III confirmatory clinical study OPTIC. The OPTIC study found that significantly more patients treated with Tepezza (82.9 percent) had a meaningful improvement in proptosis (≥ 2 mm) as compared with placebo patients (9.5 percent) without deterioration in the fellow eye at Week 24. Additional secondary endpoints were also met, including a change from baseline of at least one grade in diplopia (double vision) in 67.9 percent of patients receiving Tepezza compared to 28.6 percent of patients receiving placebo at Week 24. In a related analysis of the Phase II and Phase III clinical studies, there were more patients with complete resolution of diplopia among those treated with Tepezza (53 percent) compared with those treated with placebo (25 percent).
In October 2020, Horizon announced new long-term follow-up data from the Phase II clinical trial of Tepezza, which showed a sustained response up to one year following completion of treatment for thyroid eye disease. All patients with Week 72 data (37/37) reported some improvement in at least one of the study outcomes from baseline. 97 percent (36/37) of study participants had an improvement in clinical activity score (decrease of at least 1 point). 86 percent (31/36) had any decrease in proptosis. One patient chose elective TED surgery at Week 70 and did not have proptosis measurements at Week 72. Of patients with baseline diplopia, 70 percent (23/33) had an improvement of at least one grade. 70 percent (26/37) had disease inactivation (CAS of 0 or 1 point).
During December 2020, Horizon announced that the company expected a short-term disruption in Tepezza supply as a result of government-mandated COVID-19 vaccine production orders related to Operation Warp Speed that dramatically restricted capacity available for the production of Tepezza at its drug product contract manufacturer, Catalent. In March 2021, FDA cleared a prior approval supplement to the previously approved Biologics Licensing Application giving Horizon authorization to manufacture more Tepezza drug product resulting in an increased number of vials with each manufacturing slot. The company began to resupply the market in April, which ended the supply disruption.
 In April 2021, new pooled data from the Tepezza Phase II and III trials was published in The Lancet Diabetes & Endocrinology. This data further reinforced that Tepezza significantly improves proptosis and diplopia for TED patients in different subgroups, with most maintaining a long-term response. There was no evidence for acute disease rebound (increase in percentage of patients no longer meeting proptosis, diplopia or ophthalmic composite outcome) seven weeks after the last dose of Tepezza. Proptosis (87 percent; 62/71), diplopia (66 percent; 38/58) and ophthalmic composite outcome (92 percent; 66/72) responses were observed seven weeks after the last dose of Tepezza. A post-hoc analysis of the composite ophthalmic outcome indicated that 81 percent (68/84) of Tepezza patients versus 44 percent (38/87) of placebo patients were responders at Week 24. Proptosis (67 percent; 38/57), diplopia (69 percent; 33/48) and composite outcome response (83 percent; 48/58) were observed 51 weeks after the last dose of Tepezza for those who had long-term off-treatment data available.
In April 2021, new pooled data from the Tepezza Phase II and III trials was published in The Lancet Diabetes & Endocrinology. This data further reinforced that Tepezza significantly improves proptosis and diplopia for TED patients in different subgroups, with most maintaining a long-term response. There was no evidence for acute disease rebound (increase in percentage of patients no longer meeting proptosis, diplopia or ophthalmic composite outcome) seven weeks after the last dose of Tepezza. Proptosis (87 percent; 62/71), diplopia (66 percent; 38/58) and ophthalmic composite outcome (92 percent; 66/72) responses were observed seven weeks after the last dose of Tepezza. A post-hoc analysis of the composite ophthalmic outcome indicated that 81 percent (68/84) of Tepezza patients versus 44 percent (38/87) of placebo patients were responders at Week 24. Proptosis (67 percent; 38/57), diplopia (69 percent; 33/48) and composite outcome response (83 percent; 48/58) were observed 51 weeks after the last dose of Tepezza for those who had long-term off-treatment data available.
Additionally, in a post-hoc analysis, Tepezza-treated patients with more severe disease (those with ≥3 mm of proptosis and/or inconstant or constant diplopia) and those with less severe disease at baseline both experienced significant improvements in proptosis and diplopia. In patients with more severe disease, those treated with Tepezza had a proptosis response of 79 percent (50/63) compared to 17 percent (11/65) of those who received placebo, and a diplopia response of 68 percent (38/56) compared to 31 percent of those who received placebo (15/49). In patients with less severe disease, those treated with Tepezza had a proptosis response of 71 percent (15/21) compared to 9 percent in those who received placebo (2/22), and a diplopia response of 80 percent (8/10) compared to 30 percent in placebo (3/10).
In post-hoc analyses, patients who received Tepezza in both the lower baseline CAS subgroup (4 or 5) and the higher CAS subgroup (6 or 7) demonstrated statistically significant improvements compared with placebo in proptosis and diplopia. Overall response and CAS of 0 or 1 response also improved.
Post-hoc analysis from the Phase III study also demonstrated that in patients treated with Tepezza, those with higher (≥10 IU/L) or lower (<10 IU/L) serum thyrotropin-binding inhibitory immunoglobulin (TBII) baseline levels both had a proptosis response (mean reduction of -3.65 mm and -3.01 mm, respectively) with no treatment difference between the two groups. In patients with higher baseline TBII, 71 percent (10/14) of patients who received Tepezza experienced an improvement in diplopia compared to 23 percent (3/13) of patients who received placebo.
In November 2021, Horizon announced findings of a real-world adherence analysis of Tepezza for the treatment of TED. The analysis found that more than 90 percent (n=995) of people who were prescribed Tepezza for TED went on to complete all eight infusions, indicating a high level of adherence to the medicine in clinical practice. The study evaluated 1,101 people living with TED (71 percent female, mean age 58 years) who started treatment with Tepezza prior to July 2020. Non-compliance was low at approximately 1 percent (n=15). Only 8 percent (n=84) reported that they discontinued because of adverse events.
In June 2022, Horizon announced results of a new analysis examining rates of hyperglycemia among patients treated with Tepezza for TED compared to placebo in the Phase II and OPTIC Phase III clinical trials. The analysis found a total of nine adverse event reports of hyperglycemia in eight patients (8/84, 10 percent) who received Tepezza, and one patient (1/86; 1.2 percent) who received placebo. The majority (5/8, 63 percent) of patients who experienced hyperglycemia while taking Tepezza had pre-existing diabetes. Of the hyperglycemic AEs reported in the Tepezza-treated patients, all were controlled with medicine. All reported AEs were grade 1 (>ULN-160mg/dl) or grade 2 (161 – 250mg/dl), and none led to study discontinuation. HbA1c levels increased by 0.22 percent in those treated with Tepezza compared to 0.04 percent among placebo patients.
Ubrelvy

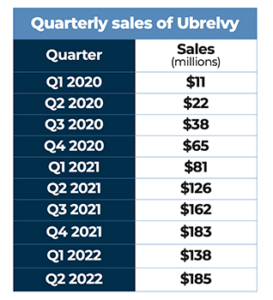
Ubrelvy was the first orally administered calcitonin gene-related peptide receptor antagonist (gepant) to be approved by FDA for the treatment of migraine attacks once they start.
Approved by FDA in late December of 2019, Ubrelvy was the first orally administered calcitonin gene-related peptide (CGRP) receptor antagonist (gepant) for the treatment of migraine attacks once they start. Ubrelvy works by blocking CGRP, a protein that is released during a migraine attack, from binding to its receptors. It works without constricting blood vessels, which some older treatments were known to do. FDA’s approval was based on four clinical studies (ACHIEVE I, ACHIEVE II, UBR-MD-04, and 3110-105-002), which demonstrated efficacy, safety, and tolerability of orally administered Ubrelvy in the acute treatment of migraine. Both 50 mg and100 mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at two hours, compared with placebo. Ubrelvy joined AbbVie’s portfolio when that company completed its acquisition of Allergan in May 2020.
In August 2020, AbbVie announced Serena Williams as the spokesperson for Ubrelvy to raise awareness of an effective acute treatment option for people living with migraine. The multichannel marketing campaign featuring Williams highlighted how Ubrelvy works for people with different lifestyles by helping individuals treat their migraine attacks anytime, anywhere. As spokesperson, she was featured in a video, available on social media, talking with neurologist and paid AbbVie consultant Dr. Jennifer McVige about her experience with migraine and Ubrelvy. Williams was also included in print and digital advertising and other marketing initiatives.
In September 2021, FDA approved AbbVie’s Qulipta, another drug from the gepant family, for the preventive treatment of episodic migraine in adults. Qulipta is the first and only oral calcitonin gene-related peptide receptor antagonist specifically developed for the preventive treatment of migraine. The approval was supported by data from a robust clinical program evaluating the efficacy, safety, and tolerability of Qulipta in nearly 2,000 patients who experienced 4 to 14 migraine days per month, including the pivotal Phase III ADVANCE study, the pivotal Phase IIb/III trial, and the Phase III long-term safety study.
 In the pivotal Phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group ADVANCE trial, the primary endpoint was change from baseline in mean monthly migraine days across the 12-week treatment period. All Qulipta dose groups met the primary endpoint and demonstrated statistically significant reductions in mean monthly migraine days compared to placebo. Patients treated with 60 mg of Qulipta across 12 weeks experienced a 4.2-day reduction from baseline of 7.8. A key secondary endpoint in the ADVANCE trial measured the proportion of patients that achieved a ≥50 percent reduction in monthly migraine days across the 12-week treatment period. The trial demonstrated that 56 percent/59 percent/61 percent of patients in the 10 mg/30 mg/60 mg Qulipta arms, respectively, achieved a 50-100 percent reduction, compared to 29 percent of patients in the placebo arm.
In the pivotal Phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group ADVANCE trial, the primary endpoint was change from baseline in mean monthly migraine days across the 12-week treatment period. All Qulipta dose groups met the primary endpoint and demonstrated statistically significant reductions in mean monthly migraine days compared to placebo. Patients treated with 60 mg of Qulipta across 12 weeks experienced a 4.2-day reduction from baseline of 7.8. A key secondary endpoint in the ADVANCE trial measured the proportion of patients that achieved a ≥50 percent reduction in monthly migraine days across the 12-week treatment period. The trial demonstrated that 56 percent/59 percent/61 percent of patients in the 10 mg/30 mg/60 mg Qulipta arms, respectively, achieved a 50-100 percent reduction, compared to 29 percent of patients in the placebo arm.
During June, AbbVie submitted a supplemental NDA to FDA for Qulipta to support the preventive treatment of chronic migraine in adults. If approved, Qulipta would be the first gepant cleared for the broad indication of the preventive treatment of migraine, including episodic and chronic. The supplemental NDA submission includes data from the pivotal Phase III PROGRESS trial in patients with chronic migraine, which supplements the existing data in episodic migraine. People living with chronic migraine experience headaches for 15 or more days per month, which, on at least eight of those days per month, have the features of migraine.
The Phase III PROGRESS trial met its primary endpoint of statistically significant reduction from baseline in mean monthly migraine days compared to placebo across the 12-week treatment period in adults with chronic migraine. The trial also demonstrated that treatment with Qulipta 60 mg once daily (QD) and 30 mg daily (BID) resulted in statistically significant improvements in all six secondary endpoints. This includes a key secondary endpoint that measured the proportion of patients that achieved at least a 50 percent reduction in mean monthly migraine days across the 12-week treatment period.
 |
Josh Slatko is contributing editor of Med Ad News and PharmaLive.com. |



{kind=link}