Diseño del estudio
El proyecto de investigación se llevó a cabo en el Laboratorio de Investigación Clínica del Instituto de Ciencias de la Salud de la Universidad de Opole, Polonia. El estudio se realizó en cooperación con el Departamento de Fisioterapia de la Universidad Médica de Wroclaw en Polonia. El Comité de Bioética local de la Universidad de Opole aprobó la investigación (n.º KB/91/FI/2018). El estudio se registró en la base de datos de registro del International Standard Randomized Controlled Trial Number (ISRCTN) (n.º ISRCTN16627714). Todos los participantes dieron su consentimiento informado por escrito para participar en el estudio. Los estudios se realizaron siguiendo la Declaración de Helsinki y las buenas prácticas clínicas.
Aleatorización y cegamiento
Los pacientes fueron seleccionados para el proyecto por un equipo de investigación formado por un internista, un radiólogo, un neurólogo, un neurocirujano, un ortopedista y un fisioterapeuta. El proyecto se diseñó como un ECA simple ciego prospectivo con observación de seguimiento. Los participantes fueron asignados al azar a grupos comparativos (experimentales o de control) mediante aleatorización simple con una proporción de 1:1 a través de una secuencia aleatoria computarizada en el sitio web random.com. La asignación al grupo fue independiente del tiempo y del personal investigador que realizaba el procedimiento. Todas las pruebas y encuestas fueron realizadas por dos fisioterapeutas, quienes entrenaron la repetibilidad y precisión de las técnicas durante 4 meses antes del inicio de la investigación. Todos los tratamientos de DN fueron realizados por un fisioterapeuta experimentado con licencia de instructor. Los ejercicios físicos fueron realizados “uno a uno” y conducidos por terapeutas certificados que también tenían 4 meses de entrenamiento por adelantado para asegurar la uniformidad del procedimiento. El personal de investigación que realizó la terapia y las mediciones no tuvo contacto con el equipo de calificación ni con quienes analizaron los resultados. El mismo terapeuta aplicó ambos procedimientos (DN y sham); sin embargo, todos los trámites se realizaron en diferentes puntos del plantel. Por lo tanto, no se pudo informar a los participantes sobre la asignación.
Tamaño de la muestra
El tamaño de la muestra del presente estudio se basó en las diferencias de los grupos en los resultados primarios (medias y desviaciones estándar de la experiencia del dolor), que se estimaron en 20 participantes (en cada grupo). Para los cálculos se permitió una disminución del 20% en el tamaño de la muestra para el seguimiento. El tamaño de la muestra se estimó en base a 10 resultados seleccionados al azar en la etapa de diseño del estudio (5 de cada grupo). Se utilizaron las medias y las desviaciones estándar de los resultados de la EVA (diferencia entre los resultados antes y después de la intervención) para estimar el tamaño de la muestra (el tamaño de muestra estimado para una prueba t de dos muestras). Parámetros del grupo experimental: media = 6,1 puntos, desviación estándar DE = 1,75 puntos; parámetros del grupo control: media = 4,4 puntos, desviación estándar DE = 1,68 puntos. El nivel alfa se fijó en 0,05 y la potencia de la prueba en 0,8. También asumió que no había correlación entre las variables evaluadas y adoptó una hipótesis nula bilateral. La estimación del tamaño de la muestra se realizó utilizando Statistica 13 (TIBCO Software Inc., Palo Alto, EE. UU.).
Participantes
Los pacientes con discopatía L5-S1 y dolor crónico (más de 3 meses), que no se sometieron a una intervención quirúrgica espinal, fueron elegibles para el proyecto. Los participantes eran mayores de edad y tenían exámenes de resonancia magnética válidos que confirmaron el diagnóstico del síndrome LBP (cambios a nivel del grado III según la clasificación Modic en la sección L5-S1). Los participantes calificados no tenían trámites previos de DN (en general, solicitudes en otros sitios).
Los criterios de exclusión fueron: ausencia de dolor y movilidad reducida en la sección lumbosacra, otras afecciones de la columna (espondilolistesis, fracturas, tumores, infecciones, enfermedades reumáticas, síndrome de cauda equina), ciática y síntomas radiculares, embarazo, marcapasos cardíaco implantado, trastornos de la coagulación sanguínea, terapia anticoagulante, terapia con esteroides, implantes metálicos dentro del sitio de aplicación, alteraciones sensoriales, trastornos mentales, cáncer, cambios en la piel dentro del sitio de tratamiento, infecciones virales y bacterianas, fiebre, agotamiento, hipertensión no tratada, tomar analgésicos y antiinflamatorios, miedo a agujas, sin consentimiento para el procedimiento.
Tratamiento
Para el grupo experimental, el estudio utilizó agujas SOMA estériles y desechables de acero quirúrgico japonés con un grosor de 0,3 mm y una longitud adaptada a la zona de punción (fig. 5). El programa completo de DN se muestra en la Fig. 6. Un solo tratamiento incluía las siguientes actividades:
-
La punción se activa en el área del surco entre las apófisis espinosas de la columna vertebral y el curso del músculo extensor dorsal. Se aplicó a engrosamientos palpables dentro del tejido blando en el segmento L1-L5 de la columna vertebral en ambos lados (el sitio de punción ligeramente por encima y lateralmente de la estructura dada). La dirección de la aguja fue espinal y caudal, con una inclinación de 30° desde la superficie de la piel hasta el apoyo de la aguja contra el cuerpo de la vértebra por debajo. La técnica de trabajo fue el “bombeo” (extensión e inserción rítmica de la aguja contra la superficie de la piel sin retirarla del todo) para estimular (inducir una respuesta de dolor). Después de algunos golpes de la aguja, hubo una pausa de aproximadamente 15 min (durante la cual no se interfirió en la aplicación) y los movimientos de la aguja se repitieron hasta el final del procedimiento o cuando desapareció la respuesta de dolor al “bombeo”. La longitud de las agujas era de 75 mm.
-
La punción se aplica dentro de bandas palpables de tejido conjuntivo dispuestas transversalmente sobre la masa del músculo extensor dorsal. Las estructuras sentidas en la sección L1-L5, en ambos lados, fueron punzonadas. La dirección de la aguja era ventral (perpendicular a la superficie de la piel) hasta sentir una resistencia característica (aprox. 1,5-2 cm de profundidad). La técnica de trabajo era torcer la aguja hacia un lado hasta que se detuviera. Luego hubo una interrupción de aproximadamente 15 min (durante los cuales no se interfirió la aplicación), y se repitieron los movimientos de la aguja hasta terminar el tiempo de tratamiento o pasar desapercibido el efecto de parada de la aguja al girar. La longitud de la aguja era igual a 30 mm.
-
La punción se activa en el neurocompartimento del nervio glúteo superior a través de punciones y la generación de respuesta de contracción local (LTR) dentro de los sitios sensibles palpables de posibles uniones del nervio glúteo superior (ambas ramas superior e inferior) con los músculos: glúteo mayor, glúteo medio, y tensor de la fascia ancha. La dirección del movimiento de la aguja era ventral (perpendicular a la superficie de la piel), con una característica sensación de densificación (dando mayor resistencia debajo de la aguja). La técnica de aplicación fue de “bombeo” a un mínimo de dos contracciones, seguido de una interrupción de aproximadamente 15 min (durante los cuales no se interfirió la aplicación) y se repitieron los movimientos de la aguja hasta el final del tiempo de tratamiento o la desaparición de la presión sistólica. respuesta al “bombeo”. La longitud de la aguja era igual a 100 mm.
-
Punción activada en el área de miogelosis del músculo en forma de pera, palpable como un engrosamiento tierno de las fibras musculares que saltan debajo de los dedos. La dirección del movimiento de la aguja era ventral (perpendicular a la superficie de la piel), con sensación de resistencia en el músculo en forma de pera. La técnica consistía en “bombear” a por lo menos dos contracciones, seguidas de una interrupción de aproximadamente 15 min (durante los cuales no se interfería la aplicación), y los movimientos de la aguja se repetían hasta el final del tiempo de tratamiento o la desaparición de la respuesta sistólica a “bombeo”. La longitud de la aguja era igual a 100 mm.
Figura 5
Equipo para trámite de DN.
Figura 6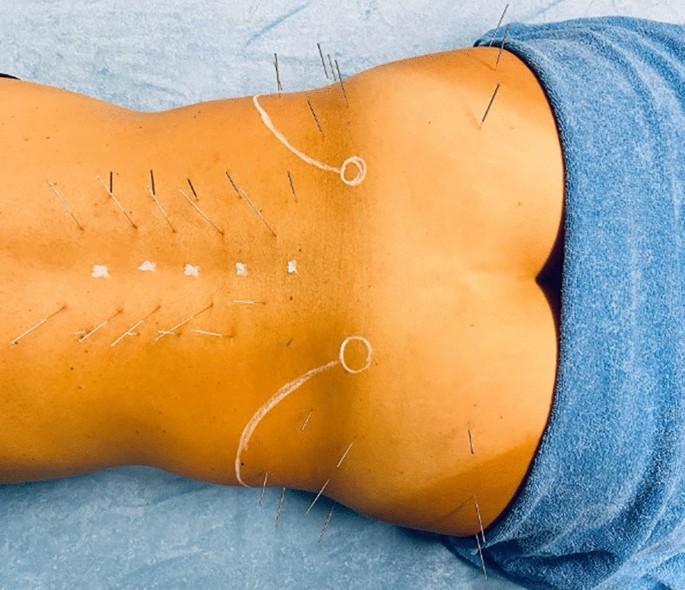
Aplicación de DN en el grupo experimental. En la figura, como puntos topográficos de referencia, se dibujan en blanco los sitios de las apófisis espinosas de las vértebras, los contornos de las crestas ilíacas y las espinas ilíacas posterosuperiores.
El grupo de control recibió terapia simulada utilizando agujas de placebo especializadas, con el diseño telescópico para colocarse sobre la piel sin perforar (ver Fig. 7). Se usaron las ubicaciones exactas de las agujas de placebo en los grupos de control y de prueba, precedidas por una palpación precisa. En puntos designados, se colocaron anillos únicos para asegurar una distancia de la piel. A continuación, los discos se fijaron con láminas finas, insertando agujas telescópicas de placebo a través de ellas. De esta forma, las agujas permanecían en la superficie de la piel sin penetrarla, permitiendo movimientos suaves para imitar la propia terapia.
Figura 7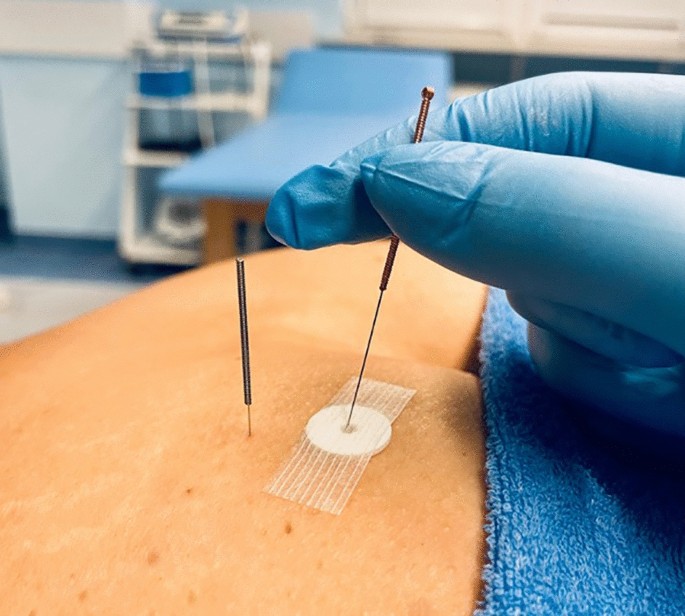
Aplicación de DN en el grupo control utilizando aguja telescópica (terapia simulada). A modo de comparación, se presenta una aguja real insertada junto al sitio de aplicación.
Los tratamientos en grupos se realizaron dos veces por semana (lunes y jueves) durante 4 semanas (8 tratamientos en total). Una sola aplicación de DN duró 60 min. Los pacientes fueron informados sobre la preparación para el procedimiento (es decir, asegurar una piel limpia, libre de ungüentos y cremas). Se utilizaron las mismas medidas asépticas en todos los grupos. Los tratamientos cumplieron con los más altos estándares de desinfección y seguridad. Los grupos también recibieron tratamientos estándar para el dolor lumbar y ejercicio físico durante un mes (45 min al día, cinco veces a la semana, de lunes a viernes). El entrenamiento de estabilización incluyó relajación miofascial de los extensores de la columna, activación del complejo lumbosacro y músculos profundos, estimulación de la respiración adecuada y activación adecuada del músculo abdominal transverso, y entrenamiento postural y dinámico. Los procedimientos se proporcionaron debido a guías clínicas recientes14,15,16.
Mediciones
Las sensaciones subjetivas de dolor se midieron mediante una escala analógica visual (VAS), en la que el paciente indicaba la intensidad del dolor de los niveles 0 a 10 (0 indica “sin dolor”, 10 indica “el dolor más intenso” del valor real) en la región LBP17. La eficiencia funcional se evaluó con el Oswestry Disability Index (ODI)18. El rango de movimiento de la columna lumbar se midió con la prueba de Schober19. En ambos grupos se tomaron medidas antes e inmediatamente después del tratamiento. Los efectos a largo plazo se midieron 1 y 3 meses después del final de la terapia20. Los participantes no utilizaron tratamientos de fisioterapia o farmacoterapia durante el período de estudio.
análisis estadístico
El análisis estadístico se realizó con el programa Statistica 13 (TIBCO, Inc., Palo Alto, EE. UU.). Para las variables medibles se calcularon medias, medianas, desviaciones estándar, cuartiles y el rango de variabilidad con valores extremos. Las variables cualitativas se expresaron en frecuencias (porcentajes). Las variables cuantitativas se analizaron con la prueba de Shapiro-Wilk para examinar la normalidad de la distribución. Las comparaciones entre grupos se realizaron con la prueba de chi-cuadrado (χ2). Las comparaciones dentro de los grupos para las mediciones 1 a 4 se realizaron mediante el análisis de varianza de Friedman y una prueba post-hoc (prueba de Dunn). Las comparaciones entre grupos (grupos experimentales frente a grupos controlados con placebo) se evaluaron mediante el análisis de varianza de Kruskal-Wallis en una prueba post-hoc (prueba de U-Mann-Whitney con corrección de Bonferroni). Se adoptó el nivel de significación de α = 0,05 para todos los análisis.
Consideraciones éticas
El protocolo de estudio fue aprobado por el Comité de Ética Local de la Universidad de Opole en Polonia (aprobación n.º KB/90/FI/2018).


